"Investigating a new respiratory virus outbreak in Santa's workshop"
Christmas eve was quickly approaching, and Santa was trying to decide whether to purchase masks for all of the reindeer again this year (he was aware that there were documented cases of SARS-CoV-2 infecting White-Tail Deer (O. virginianaus), and he knew that Reindeer (R. tarandus) were closely related), when the Chief Elf interrupted his train of thought to inform him that two of the elves in the toy workshop were in the infirmary with respiratory distress. Testing had revealed that they were both infected with Respiratory Syncitial Virus (RSV), specifically with the RSVA subtype.
Santa asked "Do we know where they were exposed, or if one of them transmitted it to the other? We may need to require everyone at the North Pole to mask again until after Christmas eve . . . "
"The diagnostic lab says the two viruses are similar but not identical. Also they are not confident of the lineage calls and the clade assignments because we are using a new set of PCR primers designed in Edinburgh, which are based on a newer RSVA reference sequence (RefSeq). The problem is that we are using the RSVA phylogenetic tree available at usher.bio and that tree was built using the NCBI RefSeq as the root. You can see where the problem arises . . . "
Santa thought for a moment and said "Oh, blast! We aligned our fastq reads to the other RefSeq, and at the primer trimming step we are using the genomic coordinates from that different virus strain. So if we first align against the NCBI RefSeq genome (to get more accurate lineage calls and clade assignments) then the primer trimming step will not work as intended. Huh! Hmmm, do you think we could align those two different RSVA RefSeqs to each other and then modify the coordinates in the primer bed file so it uses the NCBI RefSeq's genomic coordinates instead?!?"
The Chief Elf sketched this out on the iceboard and said, "I think this could work. I will ask our bioinformatic software developers to tackle the problem."
And so gentle reader that is how we arrived at a simple, but tedious task of converting the genomic coordinates in this bed file:
RS20000581 44 66 RSVA_1_LEFT 1 +
RS20000581 434 464 RSVA_1_RIGHT 1 -
RS20000581 359 385 RSVA_2_LEFT 2 +
RS20000581 749 773 RSVA_2_RIGHT 2 -
RS20000581 669 699 RSVA_3_LEFT 1 +
RS20000581 1057 1083 RSVA_3_RIGHT 1 -
RS20000581 990 1016 RSVA_4_LEFT 2 +
RS20000581 1366 1389 RSVA_4_RIGHT 2 -So that it would contain the correct coordinates from a different RSVA subtype RefSeq. It quickly became apparent that this problem could be solved much more quickly using the Awesome Power of Perl, unfortunately there was no out-of-the-box solution available in the existing base of Perl code. But how to proceed?
Using the Awesome Power of Perl in Bioinformatics
Each of the two genome files are simple text files consisting of an alphabet of five letters (A, C, G, and T (occasionally an N)) (technically RSV, similar to SARS-CoV-2 is an RNA virus, but life is simpler if we use 'T' instead of 'U'), all we need is a so-called "Global Alignment" of the two full-length sequences (they are each just over 15,000 bases in length). What we really want to know is where all of the longest stretches of identical sequences are located in the viral chromosomes. Fortunately an algorithm to calculate this was published by Needleman and Wunsch back in 1970; it is considered a classic example of dynamic programming (which builds up the answer without consuming vast amounts of memory). Furthermore, the National Center of Biotechnology Information (NCBI) maintains a server we can use to upload any two closely related nucleotide sequences.
The output from the N-W Global Alignment looks like this:
Query 2177 GTGTGATTAACTACAGTGTATTAGATTTGACAGCAGAAGAACTAGAGGCTATCAAACATC 2236
|||||||||||||||||||| |||| ||||||||||||||||||||||||||||||||||
Sbjct 2221 GTGTGATTAACTACAGTGTACTAGACTTGACAGCAGAAGAACTAGAGGCTATCAAACATC 2280
Query 2237 AGCTTAATCCAAAAGATAATGATGTAGAGCTTTGAGTTAATAAAAAGGTGGGGCAAATAA 2296
|||||||||||||||||||||||||||||||||||||||||||||| ||||||||||||
Sbjct 2281 AGCTTAATCCAAAAGATAATGATGTAGAGCTTTGAGTTAATAAAAAA-TGGGGCAAATAA 2339Where the the top sequence in each pair is a stretch of 60 bases from the Edinburgh RefSeq (Labeled 'Query') and the bottom sequence in each pair is the matching stretch from the NCBI RefSeq (Labeled 'Sbjct'). The unix pipe symbols denote a perfect match between the sequences at that base.
Notice that the numbering systems are slightly offset, AND that there is a gap introduced with a hyphen in-between the A at 2327, and the T at 2328 in the Sbjct, directly across from the G at position 2384 in the Query. That can be interpreted as a single nucleotide insertion in the Query genome. So creating the mapping table between the two coordinate systems is not trivial. The sequences only share 94 percent identity over about 15,200 nucleotides, which means they diverge 6 percent of the time.
To simplify the construction of our lookup table we can use grep to filter the starting alignment into two different subsets,
grep ^Query nw_alignment.txt > just_rows_from_edinburgh_refseq.txt
grep ^Sbjct nw_alignment.txt > just_rows_from_ncbi_refseq.txtN.B. you will have to go in and manually remove the first extraneous Query line from the first file. These become the first two input files to our script, the third file is the primer bed file we are starting with.
Our Script that Creates a Lookup Table, and then Swaps Coordinates in a bed File
#!/usr/bin/env perl
use strict;
use warnings;
my $query_input = $ARGV[0];
my $subject_input = $ARGV[1];
my $input_bed = $ARGV[2];
open my ($FH1), '<', $query_input or die "Could not open $query_input for reading\n";
open my ($FH2), '<', $subject_input or die "Could not open $subject_input for reading\n";
my @global_array = ();
$global_array[0] = { query => undef,
sbjct => undef,
};
# This global counter serves as the index for the first data structure
# that captures the information at each place in the aligned sequences
my $counter = 0;
# Parse the rows of the first file
while ( <$FH1> ) {
chomp;
# Each of these filtered rows has the same structure, in addition to
# A, C, G, and T, the N-W output can also contain hyphens (dashes), so
# our regex has to include that in the string we are capturing
my ($seq) = $_ =~ m/Query\s+\d+\s+([\w\-]+)\s+\d+/;
# Parse the 60 nucleotide string into individual characters
my @qchars = split(//, $seq);
foreach my $q ( @qchars ) {
$counter++;
# Load up the array of hashrefs with the symbols
$global_array[$counter]{query} = $q;
}
}
close $FH1;
$counter = 0;
while ( <$FH2> ) {
chomp;
my ($seq) = $_ =~ m/Sbjct\s+\d+\s+([\w\-]+)\s+\d+/;
my @schars = split(//, $seq);
foreach my $s ( @schars ) {
$counter++;
$global_array[$counter]{sbjct} = $s;
}
}
close $FH2;
# Now we process the information in each element of the
# @global_array, and store that in this global hash
# the keys of this hash are genomic coordinates for the
# Edinburgh RSVA RefSeq and the values are the genomic
# coordinates of the NCBI RSVA RefSeq
my %ncbi_coordinates_of = ();
my $qcounter = 0;
my $scounter = 0;
# There is nothing useful in the [0] element
foreach my $i ( 1.. ) {
my $key = undef;
my $value = undef;
# If the string from the N-W output contains a '-' then we
# need to handle those differently. We only increment the
# $qcounter when the value in 'query' matches a letter
if ( $global_array[$i]{query} =~ m/\w/ ) {
$qcounter++;
$key = $qcounter;
if ( $global_array[$i]{sbjct} =~ m/\w/ ) {
$scounter++;
$value = $scounter;
}
$ncbi_coordinates_of{$key} = $value;
}
else {
$scounter++;
next;
}
}
# After all of that manipulation and data wrangling, the problem now becomes
# very simple. We read in the starting primer bed file and iterate over each
# TSV record. We use the information in columns 2 and 3 to query the
# %ncbi_coordinates_of hash, and insert the coordinates we want from the hash
# values. The modified record is immediately printed to STDOUT
open my ($FH3), '<', $input_bed or die "Could not open $input_bed for reading\n";
while ( <$FH3> ) {
my @fields = split(/\t/, $_);
# Change the sequence name in column 1
$fields[0] = 'NC_038235.1';
# It is formally possible that either the 'start' or the 'end' of the
# Query sequence is NOT in the %ncbi_coordinates_of hash, so check for this
unless ( exists $ncbi_coordinates_of{$fields[1]} and exists $ncbi_coordinates_of{$fields[2]} ) {
$fields[1] = $fields[2] = 'undef';
print join("\t", @fields);
next;
}
# Sometimes the 'start' and 'end' coordinates from the Query sequence ARE keys in the
# %ncbi_coordinates_of hash, but the corresponding values from the Sbjct sequence are undef
# so continue processing gracefully when this occurs
$fields[1] = $ncbi_coordinates_of{$fields[1]} //= 'undef';
$fields[2] = $ncbi_coordinates_of{$fields[2]} //= 'undef';
print join("\t", @fields);
}
close $FH3;
exit;Conclusion
When you run the script, save the output to file, and then use grep undef to see if there were any coordinates which could NOT be successfully mapped. If there are any then you can use grep -v undef to save a copy without those lines.
What did we learn? After running this script to create the new primer bed file for the NCBI RSVA RefSeq, and then rerunning their viral sequencing pipeline to align the amplified reads to the NCBI RefSeq, the bioinformaticians working with the infirmary could see that the two isolates from the workshop elves were more closely related than they had previously thought, but since the viruses still had many mutations separating them, it seemed like these were two separate exposures (or introductions) of RSVA to the North Pole compound.
You can see that for yourself in this image (which I generated using Nextstrain's excellent auspice.us tool for visualizing phylogenetic trees):
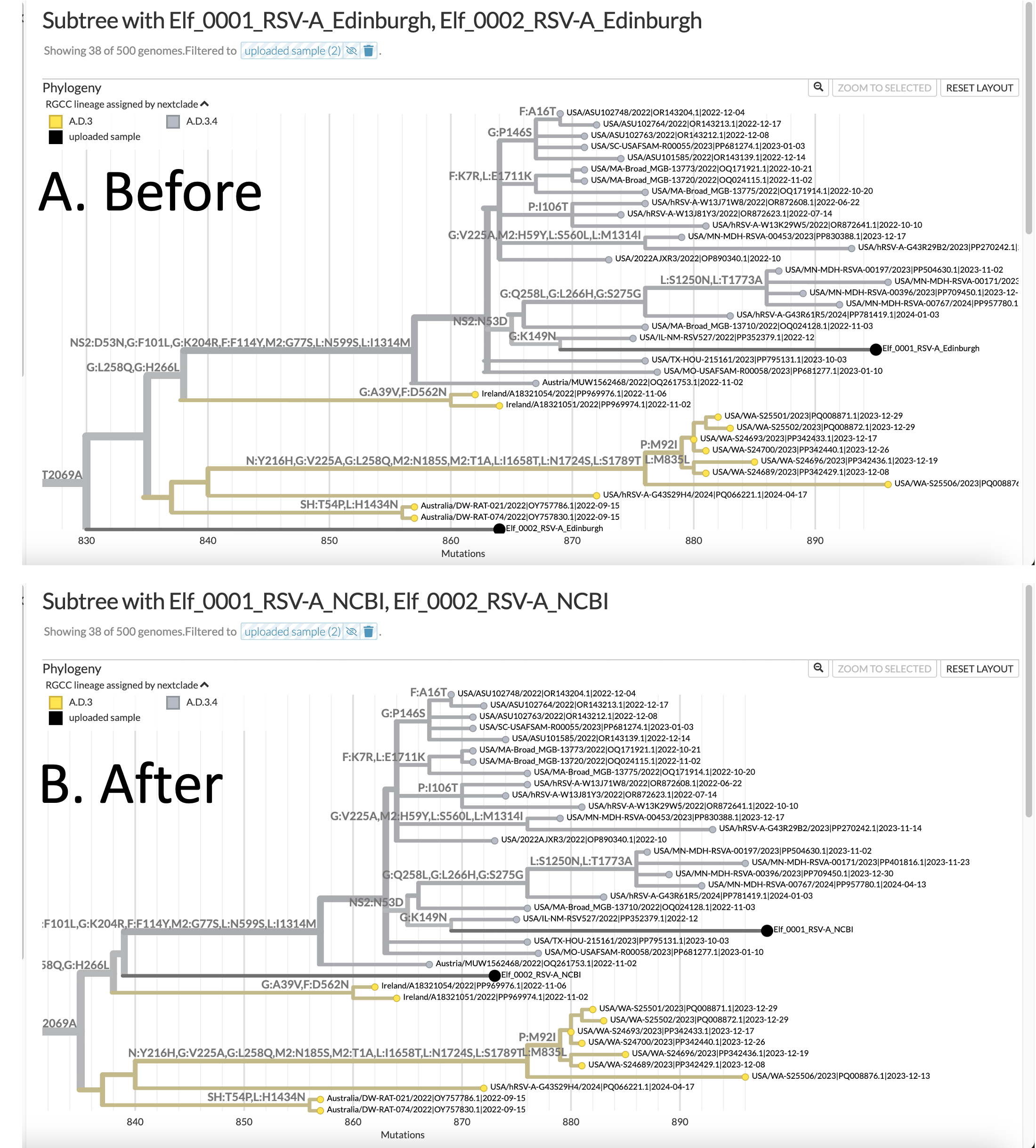
Image credit: Nextstrain.org (The UShER suite of tools for working with mutation annotated trees (MAT) has an option to generate JSON outputs pre-formatted for display at Nextstrain!)
N.B.: In accordance with the laws of Canada, Denmark, and Russia, the clinical metadata and any other personal health information (PHI) for these samples has been de-identified.
These are two versions of the RSVA Global phylogenentic tree, showing a subtree of the samples that the most highly related to the two sequences from Santa's workshop. The trees are shown rotated 90 degrees, so the "root" of the tree (which is not shown), is on the LEFT. You can see the "branches" as horizontal lines; and at the tips of each branch is a "leaf" (or a node). The leaves represent the sequences from different individual viruses in the database. Each tip is labeled with the name of the sample, and the two samples from the North Pole are colored black. The Y-axis has no units, and is arbitrary. The data depicting similarity is contained in the pattern of the branches (which are technically internal nodes), and in the LENGTH of the branches. The X-axis conveys information about the number of mutations in one sample, which is the number of nucleotide sequence changes that it takes to "get back" to the root of the tree.
Panel A. shows where the UShER program placed the two samples from the North Pole, using the Edinburgh RefSeq genome, whereas Panel B. shows where they were placed after the same samples were aligned to the NCBI RefSeq genome, and the new Primer bed file created with our script was used to remove (or trim) the primers from the end of the reads. You can see that the placement of the Elf_0001 sample is almost the same in the two trees, whereas the placement of the Elf_0002 sample has moved. It is now forking off at a different branch of the tree and is closer (and therefore more similar, or more related) to Elf_0001.
In principal, the flow and the steps used here could work with any paired RefSeqs for a pathogenic virus where you have created DNA sequencing amplicons with PCR primers from one coordinate system, but you want to be able to use your regular pipelines to align the fastq reads to a different RefSeq.
If you would like to test/replicate/hack the code above, here are links to the three input files I used:
- The RSVA Reference sequence used with the Edinburgh group's primers
- The RSVA RefSeq downloaded from NCBI (GenBank)
- The RSVA PCR Primer bed file downloaded from the Edinburgh GitHub repo
If you have some paired-end fastq files from RSV (or another RNA virus) you can get an idea of the processing steps to use to generate consensus.fasta files here.
During the COVID19 Global Pandemic these consensus.fasta files were essential for constructing the phylogenetic trees used by Epidemiologists to identify outbreaks of new strains.
THM: Best to wear masks at the North Pole until after DEC-24.
 This article contributed by: Marc Perry <marcperryster@gmail.com>
This article contributed by: Marc Perry <marcperryster@gmail.com>